Camilo García-Botero. Estudiante Doctoral en Ciencias Biológicas de la Universidad de los Andes.
Alejandro Castellanos. M.Sc. en Biología Computacional de la Universidad de los Andes.
Maria Alejandra Ulloa. Microbióloga y asistente de Investigación BCEM.
Alejandro Reyes Muñoz. PhD, profesor asociado. Departamento de Ciencias Biológicas de la Universidad de los Andes.
Cada vez que nos recuerdan que en nuestro cuerpo habitan más microorganismos que células propias [1], nos sorprendemos tanto que nos preguntamos si en realidad somos, más que humanos, un ecosistema ambulante de bacterias y otros microorganismos. Más asombroso aún es pensar que puede haber especies más abundantes que las mismas bacterias en nuestro planeta. En los últimos años, tras múltiples esfuerzos para secuenciar el ADN de distintos biomas, se ha descubierto que la abundancia de virus supera —quizás cada vez más— la de muchos otros microorganismos y se ha llegado incluso a proponer que es de un orden de magnitud superior a la abundancia de bacterias en toda la Tierra [2]. A esta gigantesca masa de nuevas entidades virales, en su mayoría de virus bacterianos conocidos como bacteriófagos, que dominan todos los ecosistemas y cuya diversidad aún permanece desconocida, se le ha denominado la «materia oscura viral» [3]
Los bacteriófagos —también llamados fagos, de cariño— no son organismos nuevos para la ciencia, pues se han estudiado desde hace décadas y han inspirado múltiples aplicaciones biotecnológicas. Quizás una de las más conocidas ha sido su uso eficaz en la lucha contra las infecciones bacterianas resistentes a antibióticos, conocida como terapia de fagos. Esta consiste en preparar cócteles de fagos especializados para atacar bacterias específicas causantes de diferentes infecciones [4].
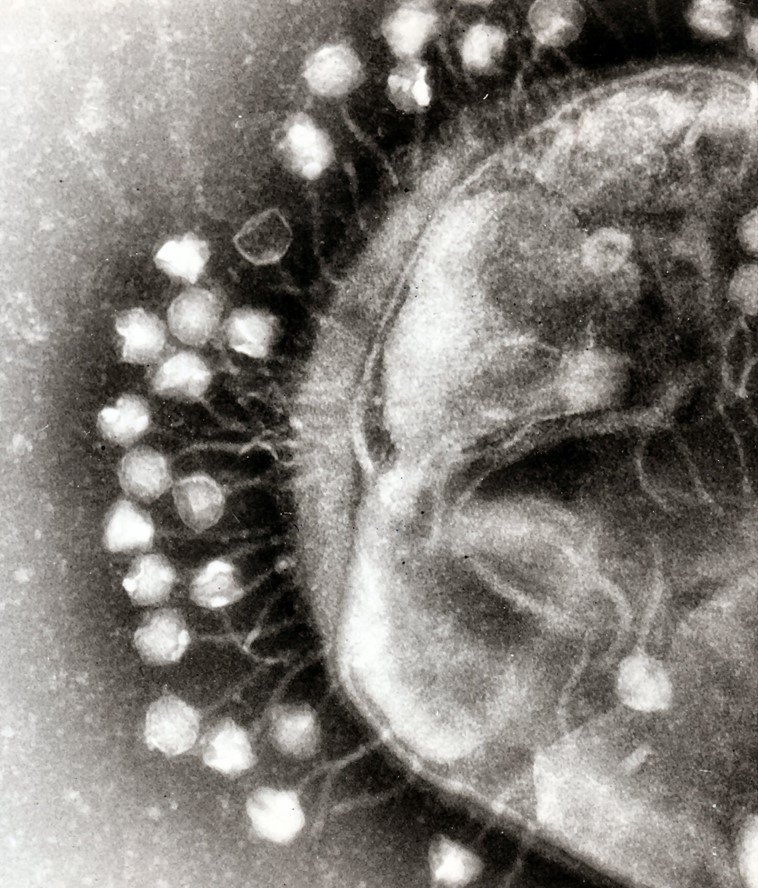
Imagen 1. Micrografía electrónica de transmisión de múltiples bacteriófagos adheridos a una pared celular bacteriana; el aumento es de aproximadamente 200 000 [9].
Se espera que conocer y develar este nuevo universo viral pueda brindar mucho más entendimiento de la vida y un sinnúmero de nuevas aplicaciones. Para esto uno de los primeros pasos fundamentales implica desentrañar su organización, sus relaciones y designar un sistema taxonómico robusto basado en las nuevas relaciones biológicas que se puedan establecer.
Ahora bien, dado que los fagos habitan en escalas casi nanométricas, es generalmente difícil observarlos y sus genomas tienden a ser pequeños también (de 1 a 500 000 pares de bases). Sin embargo, su presencia ambiental en todos los ecosistemas ha permitido que, gracias a las tecnologías de secuenciación masiva de segunda y tercera generación, en conjunto con técnicas como la metagenómica y metatranscriptómica, se puedan encontrar y ensamblar sus genomas. A este conjunto de genomas virales que no tiene cultivo u hospedero propiamente, se le conoce como UViGs [5] por sus siglas en inglés (en español, Genomas de Virus No Cultivados) y cada vez son más y más los que parece que quedan por descubrir. Solo desde el advenimiento de la secuenciación de segunda generación, en el año 2006, se han generado cerca de un millón de UViGs al 2018 [5] y ese número alcanza más de seis millones para 2023.
A pesar de que los genomas de los fagos tienden a ser pequeños, su clasificación ha sido un dolor de cabeza durante muchos años. La complejidad de esta tarea no yace en sus tamaños, sino, entre otras cosas, en que sus genomas son un mosaico de eventos evolutivos que ocurren a tasas tan rápidas que se hace difícil trazar un único ancestro para todos los taxa cultivados y no cultivados [5]. Estos últimos son los que forman —principalmente— aquella materia oscura viral.
De hecho, hace poco un consorcio de investigadores especializados en diferentes subdisciplinas de la virología se reunieron para abolir las clasificaciones virales tradicionales, cuyo enfoque se ha estado centrado en diferentes aspectos no-biológicos y han propuesto un sistema taxonómico más robusto que involucra varios principios que permitirán la expansión taxonómica en tanto se sigan conociendo más de estos nuevos virus [6, 7].
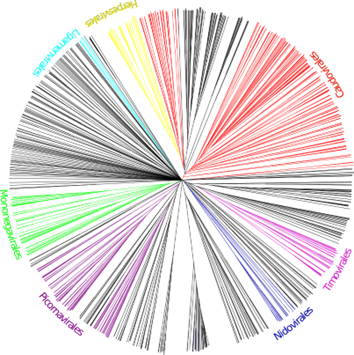
Imagen 2. Dendograma que representa la diversidad viral basado en la presencia o ausencia de ViPhOGs. Cada rama representa una secuencia viral depositada en una base de datos pública [8].
Para comprender mejor la materia oscura genética viral, desde el grupo de investigación en Biología Computacional y Ecología Microbiana (BCEM) publicamos una investigación en la que ampliamos la búsqueda de nuevas regiones conservadas en los genes virales e incluimos secuencias de virus de procariotas y eucariotas para expandir y robustecer la clasificación taxonómica viral [8]. Para lograr esto, llevamos a cabo un análisis exhaustivo de la diversidad viral almacenada en bases de datos públicas. Luego, empleamos una base de datos especializada para identificar virus y predecir las proteínas en los genomas virales que no contaban con esa información. Utilizamos tanto las proteínas ya conocidas en las bases de datos, como las predichas para identificar posibles secciones conservadas de las proteínas, llamadas dominios proteicos. Finalmente, implementamos un algoritmo de agrupamiento para definir 31150 grupos de dominios que reflejan ancestría común entre sí que nombramos ViPhOGs.
Para poner a prueba la utilidad de los ViPhOGs utilizamos un algoritmo de aprendizaje supervisado para clasificar los genomas con su respectiva taxonomía y logramos encontrar que la presencia o ausencia de ViPhOGs tiene una asociación significativa con su taxonomía [8]. Además, pudimos establecer un conjunto de 1457 ViPhOGs que, debido a su importancia en la clasificación taxonómica de los virus, podrían serconsiderados como genes marcadores asociados a distintos grupos taxonómicos definidos por el International Committee on Taxonomy of Viruses (ICTV).
Así mismo, con el objetivo de agilizar y promover el análisis de secuencias virales no solamente dentro del grupo sino en la comunidad científica, hemos estado desarrollando un flujo de trabajo computacional automatizado, amigable y robusto llamado VIRify que pueda ser utilizado por personas con limitado entrenamiento bioinformático. VIRify es capaz de identificar y ensamblar secuencias de fagos y profagos a partir de datos metagenómicos, anotar los genes de estas secuencias virales y realizar su clasificación taxonómica al utilizar los ViPhOGs.

Imagen 3. Logo del flujo de trabajo computacional para la identificación, clasificación y anotación de virus, VIRify [10].
En la actualidad, estudiantes activos del BCEM en colaboración con miembros pasados del grupo continúan con el desarrollo de nuevas aproximaciones para mejorar la clasificación taxonómica de virus a partir de nuevas aproximaciones que permitan distinguir grupos virales a distintos niveles de resolución taxonómica que sean congruentes con la historia evolutiva y la ecología del virus con relación al hospedero.
Referencias
[1] Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLOS Biol. 2016 ag.; 14(8):e1002533.
[2] Breitbart M, Bonnain C, Malki K, Sawaya NA. Phage Puppet Masters of the Marine Microbial Realm. Nat. Microbiol. 2018 jun.; 3(7):754-766.
[3] Reyes A, Semenkovich NP, Whiteson K, Rohwer F, Gordon JI. Going Viral: Next-Generation Sequencing Applied to Phage Populations in the Human Gut. Nat. Rev. Microbiol. 2012 sept.; 10(9):607-617.
[4] Kortright KE, Chan BK, Koff JL, Turner PE. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe. 2019 febr.; 25(2): 219–232.
[5] Roux S, Adriaenssens EM, Dutilh BE, Koonin EV, Kropinski AM, Krupovic M, et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019 en.; 37(1): 29-37.
[6] Turner D, Shkoporov AN, Lood C, Millard AD, Dutilh BE, Alfenas-Zerbini P, van Zyl LJ, et al. Abolishment of Morphology-Based Taxa and Change To Binomial Species Names: 2022 Taxonomy Update of the ictv Bacterial Viruses Subcommittee. Arch. Virol. 2023 febr.; 168(2): 74.
[7] Simmonds P, Adriaenssens EM, Zerbini FM, Abrescia NGA, Aiewsakun P, Alfenas-Zerbini P., et al. Four Principles to Establish a Universal Virus Taxonomy. PLOS Biol. 2023 febr.; 21(2):e3001922.
[8] Moreno-Gallego JL, Reyes A. Informative Regions in Viral Genomes. Viruses. 2021 jun.; 13(6):1164.
[9] Beards PG. Transmission electron micrograph of multiple bacteriophages attached to a bacterial cell wall; the magnification is approximately 200,000 [Internet]. Wikipedia Commons; fecha de publicación desconocida [consultado el 5 de julio del 2023]. Disponible en: https://commons.wikimedia.org/wiki/File:Phage.jpg
{kind=link}